-
And secondly the only way to really attack this in the near term (before CRISPR and like-systems) is to find out what proteins/pathways are being activated and silencing them or up-regulate them. I could fill the next 30 posts with articles and information to explain... the information is free for all to research and draw their own conclusions on the internet but in my mind AGA comes down to 4 main factors in people of European ancestry. The first is Androgenic hormones themselves. The second is the Androgen receptor. The third is overexpressed STAT3/PStat3, especially as it relates to the bulge and dermal papilla. And finally the fourth is the cytokine Interleukin-6, which is pro-inflammatory and works mainly through the JAK/STAT3 pathway. It is the interplay of these four which I think is causing the entire phenotype we see in AGA, and the correlation/association between other at risk factors in those that have AGA such as BPH, Diabetes, Prostate Cancer, Parkinson's Disease, and much greater risk of idiopathic scoliosis in females (another 20p11 major at risk spot). Stoping Ar itself will merely slow down the progression of AGA. The only way we know of so far to stop it dead in its tracks is castration, and that obviously isn't an option. I think the combination of a topical JAK/STAT inhibitor and an androgen suppressor at the same time (such as finasteride ) could potentially cause GREAT reversal in AGA phenotype. -
In my opinion this is the critical issue in AGA. What in the heck is going on at 20p11? Ar variants only confer greater risk to those of European heritage, it is monomorphic in Asians (existing in roughly only 1 type). 20p11 is by far the greatest at risk spot in asians, and is by a wide margin the second biggest risk point in Europeans. So it is the commonality between the two (along with androgenic hormones themselves... NOT androgen receptor.... only in europeans does this somehow accelerate the process).
Although this is in mice, this is a highly useful website. In postnatal mouse skin, PAX1 is primarily expressed in the dermal papilla and in a smaller amount in the dermal fibroblasts. But I think we must be careful here on this. The set of SNP's at 20p11 are between PAX1 and FOXA2. The SNP's do lie closer to PAX1... But i'm not sure in which direction the DNA is being read on this strand... aka from PAX1 to FOXA2 or from FOXA2 to PAX1. That can make a big difference. Although FOXA2 is not expressed in postnatal mouse hair follicle environment, a quick look up of the functions of FOXA2 shows that IT HAS TO BE INVOLVED SOMEHOW IN AGA. FOXA2 is involved in ANDROGEN METABOLISM and response to IL-6... you can google these if you like for proof. IL-6 has been shown to be induced by DHT in AGA dermal papilla and it increases expression through p-STAT3 in the ORS and the bulge to induce premature Catagen. Stat3 is also present in dermal papilla and in other tissues has been known to up-regulates Ar gene expression and sensitivity to Androgenic hormones. So in my mind, even though FOXA2 isn't explicitly expressed in mouse postnatal hair follicle.... FOXA2 must be involved in some way in the parthenogenesis of AGA... There is still a lot we do not know about genetics especially when it comes to "junk" DNA (The DNA not directly involved in gene expression or protein coding.... It has a function science just isn't entirely sure how to read it yet). And that is exactly what we are dealing with at 20p11 and AR/EDA2R ( At risk SNP's are between genetic coding on "junk" DNA ).Leave a comment:
-
Hmmm Calcitriol is pretty cheap t get no ?
We just need to know an amount to test....
0.1%
1%
?Leave a comment:
-
We present evidence of a new IS susceptibility locus in an ~100-kb region of chromosome 20p11.22 downstream of PAX1. Using a functional fine-mapping approach, we potentially narrow the locus to an ~1.5-kb domain with enhancer activity that is disrupted by disease-associated variants.(the IS-susceptible SNPs disrupts enhancer activity- presumably 'normal' activity for normal spinal development. We know from this study that these very same IS variants offers 'protection' from early onset baldness.) The PAX1-encoding region was originally associated with spinal development through studies of the naturally occurring undulated mouse strains. The original undulated (un) strain, first described in 1947, carries a missense mutation in Pax1 (ref. 41). Un/un mice display a curved spine with malformations of individual vertebrae including the vertebral bodies and intervertebral discs. Three additional strains, scoliosis (sco) or undulated intermediate (un-i), undulated-extensive (unex), undulated short-tail (uns), harbour partial or complete deletions of PAX1, with the latter including all of the gene and displaying the most severe phenotype41, 42.(A decrease/ablation in PAX1's expression confers malformations in the spine of rodents and amongst them- Scoliosis.) In early mouse development, Pax1 displays expression restricted to specific structures including the sclerotome that will give rise to the axial spine (vertebrae, ribs, connective tissues and skin). Genomic studies have delineated intervals downstream of Pax1 harbouring cis-regulatory activity consistent with this pattern43. In particular, transposon-based deletion mapping and reporter gene assays defined the ~148-kb region 3′ of Pax1 as necessary to drive somitic gene expression (that is, in the dorsal sclerotome) during early mouse development. Furthermore, the mouse Xe1 enhancer encoded in this region was shown to be sufficient to drive a similar expression pattern43. Our data using zebrafish transgene assays confirmed the enhancer activity of the human Xe1 orthologue and revealed another element in the region, PEC7, with potential somitic enhancer activity that was disrupted by IS-associated sequence variants.(The 'normal' variants in the affected region posseses potential somitic enhancer activity for proper spinal development, and is disrupted in those carrying the IS-associated variants.This observation strongly suggests that PEC7 itself functions in IS susceptibility, a hypothesis that may be tested in model systems by targeted mutagenesis.
All these implies that PAX1 needs to be DECREASED in order to regrow hair- because a decrease of PAX1 is what gives females a curved spine, but protection from hair loss.
But, im confused because PAX1 is involved with the profileration of stem cells
So is it an INCREASE or DECREASE of PAX1(which is expressed in the adult scalp) to regrow hair?
Any1 who can answer this question accurately has basically- found the cure to AGA because in those who have carry no other AGA-susceptibility genes(e.g AR/EDAR, HDAC9, etc locus) other than the PAX1 variant- needs only to address this gene in order to regrow hair.Leave a comment:
-

Leave a comment:
-
So tell me, because this is a critical question that i could not answer myself- whether do we need to INCREASE OR DECREASE PAX1's expression in the balding scalp(in order to regrow hair)?
Compounds that decrease PAX1:
Cacitriol(inhibits b-catenin)
Copper
Acetominophen(Panadol)
Bisphenol A(an estrogenic chemical)
Testosterone(androgenic hormone)
Compounds that increase PAX1:
Tretinoin(retinoic acid)
Valeric acid(present in anal cells of skunks)
Valproic acid(synthetic variant of Valeric acid)
Butyric acid(present in vomit)
Proprionic acid(present in sebum)
Ethinyl Estradiol(synthetic form of E2)
Curcumin
ResveratrolLeave a comment:
-
that's not what hydroxylation is and 5ar doesn't convert to 3beta-diol. 5ar converts testosterone to dht, dht is then converted by 3beta-hsd into 3beta-diol. 3beta-diol is then hydroxylated by some CP450 enzyme which i can't be bothered to research. it says that clearly in the abstract we keep posting back and forth to eachother.Leave a comment:
-
can i mix oleuropein and VPA together in one vehicle or is it best to keep them seperate. Chemical the epi vpa had a light purple coating which i just peeled off and dissolved the powder into eth/pgLeave a comment:
-
How important is it that we get some kind of AR antagonist like RU or CB that can compete with both T and DHT? It just seems like we desperately need another angle to fight androgens because raising T just seems stupid, and propecia has always felt like shooting a fly with a cannon by blowing up your endocrine system for a local problem
"ketoconazole cream 2% twice a day + finasteride + minox will cover the Androgen side if you've got good density or less aggressive AGA.
The problem with 5ar inhibitors, especially oral ones, is that they have a tendency to increase LH production and subsequently raise blood T levels and a local reduction in T's conversion to DHT leading to even more more T within cells. Suraphysical levels of T can activate the AR just as potently as DHT so it just offsets the potential gain from DHT inhibition. It's a tricky decision, yes Dut will kill more DHT, but you'll have even more T in comparison to Fin. I would stick to Fin."Leave a comment:
-
After looking around further, I think it's most likely PAX1. There are PAX1 enhancer regions downstream of PAX1.
A PAX1 enhancer locus is associated with susceptibility to idiopathic scoliosis in females
Idiopathic scoliosis (IS) is a common paediatric musculoskeletal disease that displays a strong female bias. By performing a genome-wide association study (GWAS) of 3,102 individuals, we identify significant associations with 20p11.22 SNPs for females (P=6.89 � 10−9) but not males (P=0.71). This association with IS is also found in independent female cohorts from the United States of America and Japan (overall P=2.15 � 10−10, OR=1.30 (rs6137473)). Unexpectedly, the 20p11.22 IS risk alleles were previously associated with protection from early-onset alopecia, another sexually dimorphic condition. The 174-kb associated locus is distal to PAX1, which encodes paired box 1, a transcription factor involved in spine development. We identify a sequence in the associated locus with enhancer activity in zebrafish somitic muscle and spinal cord, an activity that is abolished by IS-associated SNPs. We thus identify a sexually dimorphic IS susceptibility locus, and propose the first functionally defined candidate mutations in an enhancer that may regulate expression in specific spinal cells.IS is a sexually dimorphic disease10. Girls and boys exhibit a striking difference in the prevalence of progressive IS, with girls having approximately tenfold greater risk of progressive curves that require operative treatment11. This dichotomy in female/male disease expression, and its correlation with the adolescent growth spurt have prompted investigations of hormonal influences in the development and progression of female IS6.To discover new genetic risk factors for IS, we performed a two-stage GWAS in 3,102 individuals. Our results define a new susceptibility locus encoding associated SNPs that, surprisingly, are also associated with androgenic alopecia (AGA), or male pattern baldness. We find that the locus is specifically associated with female IS, suggesting that it contributes to the sexually dimorphic expression of the disease. By functional fine-mapping assays in zebrafish, we further define a sequence in the associated locus with enhancer activity that is abolished by IS-associated SNPs. Altogether, our results identify the first functionally characterized candidate mutations for IS susceptibility and expand our understanding of the role of non-coding regulatory elements in the disease. Our findings also suggest hypotheses to explain disease pathogenesis and provide the first insights into its puzzling sexual dimorphism.Comparing our results to the National Human Genome Research Institute (NHGRI) GWAS catalogue22, we found that the chromosome 20 IS locus was previously associated with early-onset male pattern baldness (AGA). Similar to IS, AGA displays sexual dimorphism, that is, it is biologically unequal in males and females. However, unlike IS, disease progression in AGA (extent of hair loss) is generally more severe in males than in females36. We identified chromosome 20p11.22 SNPs that were previously associated with AGA and that were genotyped in our GWAS37, 38, 39. In this comparison, SNPs that were associated with IS and AGA displayed the opposite direction of effect for the two disorders (Supplementary Table 1). This observation suggested that sequences in the region conferring susceptibility to IS have a protective effect in AGA. To test whether the association we observed was sex-specific, we re-evaluated association with SNPs in the 20p11.22 locus after stratification by sex, that is, separating males and females. This analysis yielded evidence for association with IS in females but not males, with a combined Fisher�s P=6.88 � 10−9 in the former data set (Table 1 and Supplementary Tables 2 and 3).Our investigation of the chromosome 20p11 locus provides the first genetic evidence to explain the puzzling sexual dimorphism that is a hallmark of IS. Besides susceptibility to progression, the pattern, onset and flexibility of deformity also differ between boys and girls10. Various hypotheses have been proposed to explain male/female differences in IS, including the existence of X-linked genetic risk factors and effects on circulating hormones. Neither mechanism has been clearly supported, although investigations have been limited6, 58. Our identification of a female-specific IS susceptibility locus suggests an underlying mechanism that is sensitive to the female milieu at the time of adolescence. Although we did not find evidence for oestrogen receptor-binding sites within the PEC7 enhancer locus itself, it is interesting to postulate that this locus increases risk of IS via downstream hormonal interactions. We note in this regard that the next-nearest gene, FOXA2, is implicated in sexually dimorphic gene expression via cooperation with androgen and oestrogen receptor59. It is possible that PEC7 regulates FOXA2. However, we did not detect Foxa2 expression in embryonic or postnatal mouse spine (data not shown) and consider it an unlikely candidate for IS susceptibility. PAX1 is also expressed in the adult scalp37. Whether variants in PEC7 affect this expression and drive association with early-onset male pattern baldness requires further study, but the overlapping genetic association suggests a possible correlation between the two sexually dimorphic conditions.Leave a comment:
-
Reponse desired
Thanks for the great write up first off!
Secondly is there anything I can do without fin/minox? wanting kids soon and had a very bad reaction to minox (not itching but skin ageing).
Many thanks,Leave a comment:
-
-
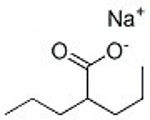
I am mainly working with VPA since that was what I first obtained, and I've got this bottle of it and don't want to waste it. I got the VPA before thinking through VPA vs. SV issues. Maybe I'll someday replace the VPA with SV, but the formulas I'm working on are complicated, and once I get the bugs worked out with VPA, I'll probably stick with that.
Take those product sheets with a grain of salt. The pharmaceutical lab I work in happened to have a small amount of SV, which I discreetly used for a couple solubility tests. The product sheet you link to says SV goes to 3% in ethanol, but I put SV in ethanol at 8%, and it was very easily soluble. Another example, Lithium Chloride that I dissolved in ethanol was only half as soluble as the numbers reported.
In solution, VPA and SV will be chemically identical. Take a look at the two structures side-by-side. You'll see that the only difference is in one single spot where VPA has a hydrogen (H) and SV has sodium (Na). In solution, that H, and that Na, float away (dissociate). All the other bonds in the molecule are permanent (covalent) bonds, and so those atoms continue to stick together. The sodium weighs more than the hydrogen though, so when you take that into account, that's why 8.3% SV will be equivalent to 7.2% VPA, the same number of molecules of valproate per volume of liquid. Also, the H that dissociates from VPA is what makes it an acid. I had concerns about the acidity, but those theoretical concerns turned out to be a non-issue.Leave a comment:
-
Well, I could be wrong, I guess. I've been wrong about many things before, and I'll be wrong about many things in the future.
Speaking of genetics, I've been playing around with something called RegulomeDB today. You put in a SNP or list of SNPs, and it combs through high-throughput experimental data and shows you epigenetic factors that those SNPs could be affecting. I threw in all the SNPs in Table S4 from this study, but focused mostly on the X-chromosome location (EDA2R/AR, with a bunch of SNPs in linkage disequilibrium) and the Chr20 location (around a long non-coding RNA segment called LINC01432 between PAX1 and FOXA2, again with a bunch of SNPs in linkage disequilibrium). The interesting thing about these two is that having the risk alleles for both can act synergistically to give you a 7x risk rather than the 2.8x or whatever it is you would get if you multiplied the odd ratios.Originally posted by Chemical
So...The strongest hit in the EDA2R/AR region is from rs6152 on AR, which is just under 1.7kb downstream of the AR promoter: http://regulome.stanford.edu/snp/chrX/66765626
You can see that EZH2 is there, so maybe the SNP affects EZH2 binding affinity. Since EZH2 is a methyltransferase, maybe rs6152 = A is more highly methylated (and therefore transcription of AR repressed). It's in a good spot for that too. See Fig1a: Negative correlation between methylation and expression is enriched in extensive regions downstream of promoters.
For the Chr20 locus, the highest score was with rs6137444 (position chr20:21785638), the most upstream of the Chr20 SNPs associated with AGA: http://regulome.stanford.edu/snp/chr20/21785638
As you can see, it's around an AR binding site. It probably has nothing to do with FOXA2, since FOXA2 isn't expressed at all in hair follicles. It may affect PAX1, which is expressed in DP cells and which plays a role in pattern formation. Or maybe the culprit is the long non-coding RNA -- long non-coding RNA can have epigenetic roles. Of note:
So maybe androgen-dependent AR binding to this site slowly and permanently changes the epigenetic program of DPCs, which could be irreversible even with AR knockdown. Who the hell knows? It's fun to speculate.Noncoding transcription has even been shown to induce the formation of heterochromatin at the p15 tumor suppressor gene locus that persisted after noncoding transcription was turned off, suggesting that the transient expression of ncRNAs can have long-lasting heritable effects on gene expression (Yu et al. 2008).
It would be an interesting experiment if some researcher could see what happens when LINC01432 is overexpressed vs. underexpressed, what roles it may have, whether AR increases or decreases its expression in the presence or absence of DHT, and so on. And then maybe do the same with PAX1.
Of course, these may not actually be the functional variants, but it's fun to play around with this stuff even with limited data.
I just Googled "site-specific gene therapy", and it seems to be a topic of ongoing research.Originally posted by Chemical
But these are specialized adult stem cells, so they'll be programmed a particular way, right?Originally posted by Chemical
Can you give some examples of people who stopped responding to RU? I can't explain it (if they're using real RU and it hasn't degraded).Originally posted by Chemical
Yes, I will say that I trust some sources more than others, but I won't get into that here.Originally posted by Chemical
The interesting thing is that in AGA males, beta-catenin binding to AR is more enriched with Wnt signaling alone than with DHT alone. They do use a small DHT concentration though, so I imagine that could change with higher concentrations. Nevertheless, it seems like beta-catenin is still able to bind TCF/LEF even when bound to AR (the luciferase activity is high in AGA40M after Wnt despite the AR/beta-catenin binding stain being pretty dark). However, AR could still maybe act as a co-transcription factor at Wnt target genes and screw things up.Originally posted by Chemical
By the way:
Acetylation of β-Catenin by p300 Regulates β-Catenin-Tcf4 Interaction
Lysine acetylation modulates the activities of nonhistone regulatory proteins and plays a critical role in the regulation of cellular gene transcription. In this study, we showed that the transcriptional coactivator p300 acetylated β-catenin at lysine 345, located in arm repeat 6, in vitro and in vivo. Acetylation of this residue increased the affinity of β-catenin for Tcf4, and the cellular Tcf4-bound pool of β-catenin was significantly enriched in acetylated form. We demonstrated that the acetyltransferase activity of p300 was required for efficient activation of transcription mediated by β-catenin/Tcf4 and that the cooperation between p300 and β-catenin was severely reduced by the K345R mutation, implying that acetylation of β-catenin plays a part in the coactivation of β-catenin by p300. Interestingly, acetylation of β-catenin had opposite, negative effects on the binding of β-catenin to the androgen receptor. Our data suggest that acetylation of β-catenin in the arm 6 domain regulates β-catenin transcriptional activity by differentially modulating its affinity for Tcf4 and the androgen receptor. Thus, our results describe a new mechanism by which p300 might regulate β-catenin transcriptional activity.The better we understand it, the better chance we have of being able to do that, I guess. We'll be better able to predict the outcome of manipulating certain things.Originally posted by Chemical
An interesting note about TGF-beta from the Tasseff paper I posted a few months ago:Originally posted by Chemical
We investigated the possibility that inhibitory signaling genes may be in the DP enriched group identified as LFO cluster 2, but not well described by the static intracellular expression model. We expect such signaling genes to display an increased expression 14 to 16 days after morphogenesis, near the on-set of catagen and before the sharp decline in the expanding population (Figure 4). Using this criterion, we identified 88 expression signals (relating to 74 unique genes; see Supplementary File S4). We observed that these expression signals, on average, are consistent with population driven changes until near catagen on-set, where they begin to increase more than what was explained by static intracellular expression assumed in the 2-population model (Supplementary Figure S10). Of these genes, 50 were annotated as extracellular genes which yields and enrichment p-value of 7.46E-18, improved enrichment over cluster 2 with a p-value of 3.63E-8. For a full list of significant enrichment categories see Supplementary File S5. Interestingly, this relatively short list includes Tgfβ2, which is currently thought to be one of the signaling molecules produced in DP cells to initiate apoptosis in hair epithelial cells at catagen on-set [7].
Given the observed expression signal, membership in DP enriched cluster 2, high enrichment for extracellular genes and inclusion of Tgfβ2, this list may contain potential targets for molecules that communicate an inhibitory signal from the DP to proliferating hair epithelial cells, closing a negative feedback loop. Obviously further experiments will be required to test this hypothesis; however, it does provide a starting point for future validation of the conclusions drawn above and, perhaps, even those identified in the model of Al-Nuaimi et al. [10].Probably some of the DKK1 is by the ROS -> p53 -> DKK1 pathway you pointed out (there's a p53 binding site on the DKK1 promoter), but also that tail-off effect is really similar to prostate cancer graphs/stain patterns of Wnt target genes with increasing concentrations of DHT, and DKK1 is a direct Wnt target gene. Adding DHT could possibly translocate beta-catenin to the nucleus if it's bound to AR and can ride AR into the nucleus.Originally posted by ChemicalLeave a comment:















Leave a comment: