
 Tweet
Tweet
Hi all, I've been doing some research on MBP for the pat few weeks and I thought I'd share some of the interesting insights on this topic, its a long read but you can skip the abstracts and articles for my conclusions. Please do not hesitate to ask questions and I strongly encourage people to dispute this research, as this will lead to a beneficial discussion on points that I havent considered.
Lets look at how DHT works to cause MBP - in depth.

Here are some studies in support of the DHT metabolite 3Beta diol being a potent activator of Estrogen Receptor Beta.
5α-Androstane-3β,17β-diol (3β-diol), an estrogenic metabolite of 5α-dihydrotestosterone, is a potent modulator of estrogen receptor ERβ expression in the ventral prostrate of adult rats
An endocrine pathway in the prostate, ERβ, AR, 5α-androstane-3β,17β-diol, and CYP7B1, regulates prostate growth
Back to the original article on DHT�s effect on the prostate.
It appears VEGF expression is crucial for hair follicle anagen induction and maintenance as described by this study:
This shows that VEGF is crucial for hair follicle survival and a reduction in VEGF mediated angiogenesis (formation of new blood vessels to supply hair follicles) could prevent the transition from telogen to anagen.
Heres a study demonstrating the role of hif-1 in VEGF regulation:
Androgens actually enhances the expression of HIF-1 via Androgen Receptor mediated signalling which explains the procarcinogenic effect of androgens in prostate cancer:

This could explain the vellus to terminal transition of follicles all over the body in males, especially facial hair. We can conclude from this that it is indeed possible for vellus hair follicles to become terminal given the right environmental conditions (local growth promoting agents) and time. Males require a significant number of years of exposure to androgens to develop thick facial and body hair.
Evolution could be responsible for this DHT driven inhibition of androgen's procarcinogenic effects i.e the males with higher androgens were physically and mentally (androgen increases grey matter/spatial ability) stronger but were also susceptible to early cancer related deaths, so the males that ended up surviving were the ones that had the AR related edge and developed a feedback loop that canceled out the negative effects of androgens. Some of these biological feedback loops have been refined over thousands of years an as a result have become increasingly complex due to natural selection (survival of the fittest/most adaptable).
This is all interesting with regards to the prostate but what about the scalp?
First lets take a look at how exactly 3 beta diol is synthesized.
This study is incredibly crucial to understanding the regional differences in Androgen metabolism and how the hairline is affected the most. We know that 5ar is elevated in balding regions but furthermore, the enzymes responsible for catalyzing the conversion of DHT to 3beta diol are also elevated in alopecic regions, whereas the enzymes responsible for synthesizing anrostanedione were higher in non alopecic regions.
When I read this I immediately recalled a compound that is commonly used for treating Androgenetic Alopecia: Ketoconazole (nizoral).
Ketoconazole is actually a really effective inhibitor of androgen synthesis, and a weak inhibitor of Androgen receptor.
3 beta-hydroxy-5-ene-steroid oxidoreductase = 3 beta HSD
So for clarification:
Ketoconazole/miconzole inhibit 3 Beta HSD
3 beta HSD converts DHT to 3beta diol.
3beta diol activates ERbeta.
ERbeta destabilizes HIF-1 and results in loss of VEGF expression.
Hair follicles fail to enter anagen due to lack of bood supply.
This is a potential explanation of the mechanism behind ketoconazoles hair growth/hair loss prevention effects.
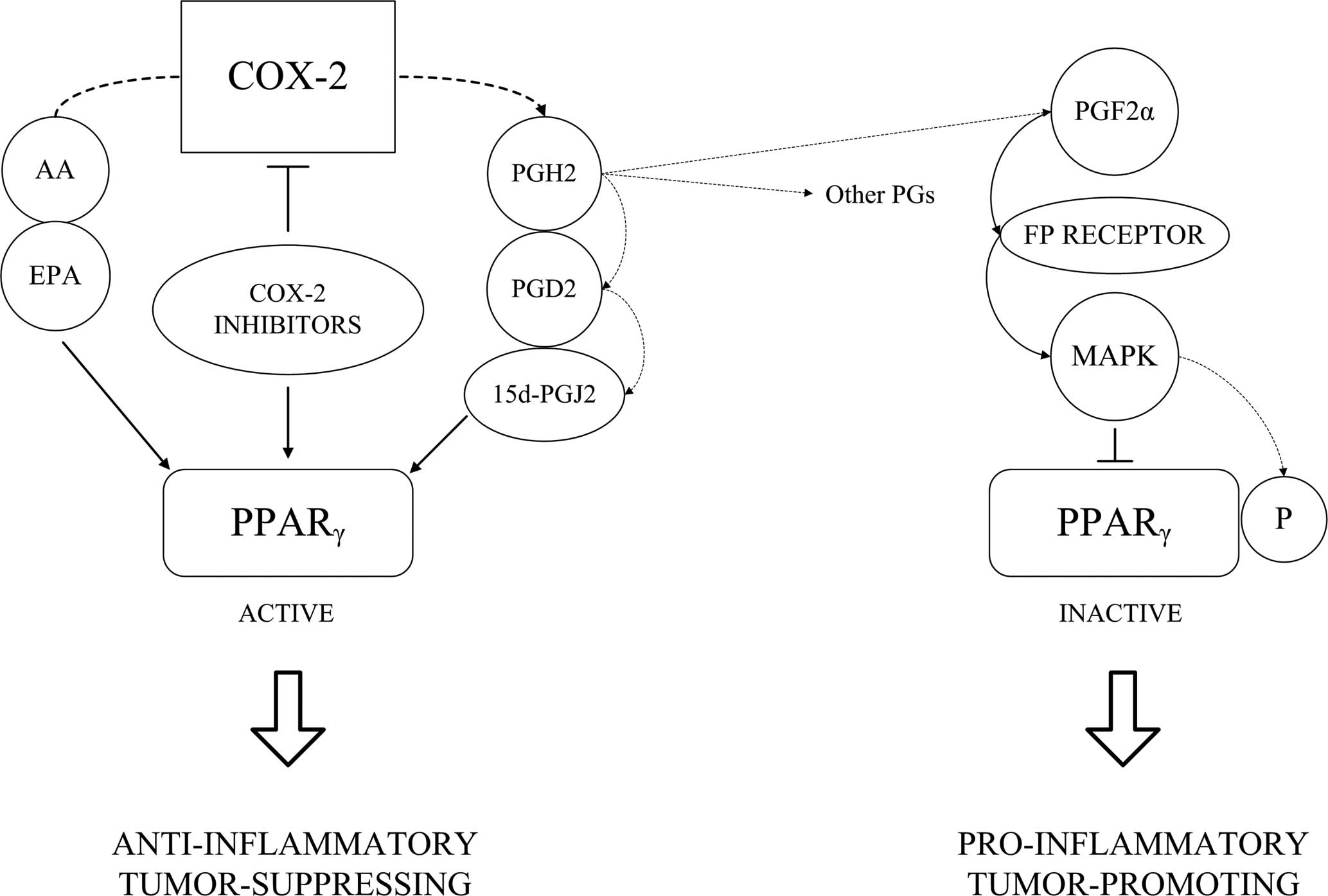
I also wanted to talk a bit about PGD2. Specifically it�s metabolite:

I�m still doing research on this by product of PGD2 as it may hold more relevance than PGD2 itself.
Another very crucial effect of DHT is the elevation Dickkopf-1 (DKK1).
So how exactly does dkk1 work?
Lets take a look at the importance of LRP6.

So without LRP6, the canonical WNT pathway cannot be activated. This means Beta Catenin will be degraded by GSK3.
It�s very clear that without Beta Catenin hair will not form:
The real question is how does DHT actually increase DKK1? This is something that I�ve been struggling to figure out. Finding an effective and feasible way to prevent the increase of DKK1 as a result of DHT may allow us to focus our attention away from DHT inhibitors, and also allow other hair promoting agents to work better (although even without agonists the body�s natural wnt proteins can have a chance to bind to the LRP6 and do their job).
I�ve come across mainly dead ends and no studies to date actually document how Dkk1 is regulated.
We know that L-ascorbic acid 2-phosphate and L-threonate, an ascorbate metabolite inhibits DHT induced DKK-1.
Dkk-1 is also induced by p53: http://www.ncbi.nlm.nih.gov/pubmed/10777218
DHT increases p53 and p21 significantly: http://www.ncbi.nlm.nih.gov/pubmed/22859066
This is a weak connection at best.
Other possible links include:
It appears VDR (Vitamin D receptor) mediates DKK-1 protein expression in the presence of E cadherin by binding to a promoter region on Dkk1 gene.
Furthermore, DHT has been shown to upregulate VDR via - wait for it � ERBeta!
Although this study�s focus is on breast cancer cells, it�s still points us in the right direction.
Vitamin D has been shown to have a biphasic effect on hair growth:
At higher concentrations of 1,25(OH)2D3, there was a dose-dependent inhibition of both follicle and fiber growth (IC50 values of 100 nM), in part due to reduction in the growth periods. There was a marked delay between the onset of 1,25(OH)2D-induced hair follicle and hair fiber growth inhibition.
This exactly inline with the oxford study:
So to conclude, it�s quite apparent there are a multitude of factors involved in the prognosis of MBP, but certain pathways have been overlooked which I believe has stagnated potential progress.
I strongly believe that inhibiting DKK-1 or reducing it will enable other treatments to work far more effectively, along with mitigating the detrimental effects of downstream DHT pathways i.e the actual effect instead of the overall mediator. The answer lies in the details, we must fully understand the mechanisms � the how and why before trying to exploit the pathway for our own gain. This is the key to hacking and efficient problem solving, you find the weakest link and plan your attack on that.
In the next post I will discuss therapeutic applications of these findings and some experiments I�ve been conducting (with interesting results), after all, what good is theory without application?
Lets look at how DHT works to cause MBP - in depth.
The Anticancer Testosterone Metabolite 3β-Adiol
Since DHT is generally agreed to be carcinogenic, the thought was that reducing the transformation of testosterone into DHT would reduce cancer risk. In the Prostate Cancer Prevention Trial (PCPT), 18,882 men over age 55 years with normal prostate examinations and a PSA below 3.0 were randomly assigned finasteride or placebo for seven years.
Slightly Lower Cancer Risk, Much Higher Killer Cancer Risk
The results were a surprise to the researchers, who found � as they expected � a lower rate of cancer in the finasteride group than in the placebo group (18.4% versus 24.4%). What surprised them was the considerably higher number of significantly aggressive cancers � for the technically inclined, higher Gleason scores � among those in the finasteride group with cancer versus those with cancer in the placebo group (37% versus 22.3%). That�s why you never saw a television commercial about finasteride preventing prostate cancer. To be accurate it would have to say, �Take finasteride! It lowers your risk of prostate cancer, but if you do get prostate cancer, you�re more likely to die of it!�
The PCPT researchers concluded, �finasteride prevents or delays the appearance of prostate cancer, but this possible benefit and a reduced risk of urinary problems must be weighed against sexual side effects and the increased risk of high-grade prostate cancer.� A separate meta-analysis published as a Cochrane Review found that 5a-reductase inhibitors, including finasteride, have inadequate evidence to say that these patent medicines reduce mortality, in terms of prostate cancer.
It�s true that if DHT alone is considered, elevated levels of DHT might be thought to increase your prostate cancer risk, as DHT is a procarcinogenic (for the technically inclined, dedifferentiating) meta�bolite.
However, thanks to evolution and Nature has developed a natural way of counter acting the procarcinogenic effects of DHT! If we take the metabolites of DHT into consideration, too, elevated DHT may or may not have this effect. One of these metabolites may actually offset or even reduce any DHT-increased risk. How does that happen?
After testosterone is converted to DHT, DHT is in turn normally metabolized into a relatively smaller quantity of 5a-androstane-3a,17b-diol (abbreviated as 3a-Adiol), and a usually larger amount of 5a-androstane-3b,17b-diol (abbreviated as 3b-Adiol). These same researchers also report that while nearly all the 3a-Adiol is converted back to DHT (which presumably makes 3a-Adiol a �pre-procarcinogen�), the 3b-Adiol does not convert back to 5a-DHT. Very importantly, they report that 3b-Adiol is an anticarcinogen (for the technically inclined, a redifferentiating agent) that activates estrogen receptor beta, an anticarcinogenic estrogen receptor present in large numbers in the prostate gland.5 (Estrogen receptor beta is present in many other tissues in both sexes, but that�s a topic to be explored at another time.)
Since DHT is generally agreed to be carcinogenic, the thought was that reducing the transformation of testosterone into DHT would reduce cancer risk. In the Prostate Cancer Prevention Trial (PCPT), 18,882 men over age 55 years with normal prostate examinations and a PSA below 3.0 were randomly assigned finasteride or placebo for seven years.
Slightly Lower Cancer Risk, Much Higher Killer Cancer Risk
The results were a surprise to the researchers, who found � as they expected � a lower rate of cancer in the finasteride group than in the placebo group (18.4% versus 24.4%). What surprised them was the considerably higher number of significantly aggressive cancers � for the technically inclined, higher Gleason scores � among those in the finasteride group with cancer versus those with cancer in the placebo group (37% versus 22.3%). That�s why you never saw a television commercial about finasteride preventing prostate cancer. To be accurate it would have to say, �Take finasteride! It lowers your risk of prostate cancer, but if you do get prostate cancer, you�re more likely to die of it!�
The PCPT researchers concluded, �finasteride prevents or delays the appearance of prostate cancer, but this possible benefit and a reduced risk of urinary problems must be weighed against sexual side effects and the increased risk of high-grade prostate cancer.� A separate meta-analysis published as a Cochrane Review found that 5a-reductase inhibitors, including finasteride, have inadequate evidence to say that these patent medicines reduce mortality, in terms of prostate cancer.
It�s true that if DHT alone is considered, elevated levels of DHT might be thought to increase your prostate cancer risk, as DHT is a procarcinogenic (for the technically inclined, dedifferentiating) meta�bolite.
However, thanks to evolution and Nature has developed a natural way of counter acting the procarcinogenic effects of DHT! If we take the metabolites of DHT into consideration, too, elevated DHT may or may not have this effect. One of these metabolites may actually offset or even reduce any DHT-increased risk. How does that happen?
After testosterone is converted to DHT, DHT is in turn normally metabolized into a relatively smaller quantity of 5a-androstane-3a,17b-diol (abbreviated as 3a-Adiol), and a usually larger amount of 5a-androstane-3b,17b-diol (abbreviated as 3b-Adiol). These same researchers also report that while nearly all the 3a-Adiol is converted back to DHT (which presumably makes 3a-Adiol a �pre-procarcinogen�), the 3b-Adiol does not convert back to 5a-DHT. Very importantly, they report that 3b-Adiol is an anticarcinogen (for the technically inclined, a redifferentiating agent) that activates estrogen receptor beta, an anticarcinogenic estrogen receptor present in large numbers in the prostate gland.5 (Estrogen receptor beta is present in many other tissues in both sexes, but that�s a topic to be explored at another time.)
Here are some studies in support of the DHT metabolite 3Beta diol being a potent activator of Estrogen Receptor Beta.
5α-Androstane-3β,17β-diol (3β-diol), an estrogenic metabolite of 5α-dihydrotestosterone, is a potent modulator of estrogen receptor ERβ expression in the ventral prostrate of adult rats
An endocrine pathway in the prostate, ERβ, AR, 5α-androstane-3β,17β-diol, and CYP7B1, regulates prostate growth
Back to the original article on DHT�s effect on the prostate.
In a letter to the editor of the New England Journal of Medicine, Otabek Imamov, MD, et al. state, �[DHT] is the fulcrum in this balance. It suggests that finasteride, by blocking the conversion of testosterone to [DHT], inhibits the production of [3b-Adiol] thus suppressing [the anticarcinogenic activity of] ERb and preventing the [re]-differentiation of epithelium. This mechanism could account for the higher incidence of poorly differentiated tumors in the finasteride group in the Prostate Cancer Prevention Trial.�6
A review in the Biology of Reproduction Journal states, �We believe that a higher incidence of low-differentiated [more aggressive] tumors in the finasteride-treated arm observed in the PCPT is caused by altering the normal differentiation of prostatic epithelium in the environment lacking the natural ERb ligand � [3b-Adiol].�7
Research has found some very specific things that 3b-Adiol does to inhibit prostate cancer growth. According to the title of a 2005 research report: �The androgen derivative 5a-androstane-3b, 17b-diol [3b-Adiol] inhibits prostate cancer cell migration through activation of the estrogen receptor beta subtype.�8 Other researchers reported that �3b-Adiol not only inhibits PC3-Luc cell [a specific type of prostate cancer cell] migratory properties, but also induces a broader antitumor phenotype [type of cell] by decreasing the proliferation [growth] rate, increasing cell adhesion [cancer cells don�t �stick� as normal cells do] and reducing invasive capabilities in vitro.�9 But these researchers went beyond test-tubes to living mice, writing �In vivo, continuous administration of 3b-Adiol reduces growth of established tumors and counteracts metastasis formation when PC3-Luc cells are engrafted subcutaneously in nude mice or are injected into the prostate.�
The conclusion to this research article was very encouraging: �Since 3b-Adiol has no androgenic activity, and cannot be converted to androgenic compounds, the effects here described entail a novel potential application of this agent against human PC.�9 A novel potential application of 3b-Adiol, a totally natural human testosterone, against human prostate cancer! Where are the headlines? This article was published in 2010!
For the really technically inclined, here are several �mechanisms of action� of 3b-Adiol, all of which come from stimulation of estrogen receptor beta:
- repression of VEGF-A (vascular endothelial growth factor A) expression
- destabilization of HIF-1a (hypoxia-inducible factor 1a)
- reduction of �Snail1″ relocation from the cytoplasm to the nucleus of cancer cells
According to the researchers who published the above mechanisms of action, �� high Gleason grade cancers � exhibit significantly more HIF-1a and VEGF-A and Snail1 nuclear localization compared to low Gleason grade cancers.�
A review in the Biology of Reproduction Journal states, �We believe that a higher incidence of low-differentiated [more aggressive] tumors in the finasteride-treated arm observed in the PCPT is caused by altering the normal differentiation of prostatic epithelium in the environment lacking the natural ERb ligand � [3b-Adiol].�7
Research has found some very specific things that 3b-Adiol does to inhibit prostate cancer growth. According to the title of a 2005 research report: �The androgen derivative 5a-androstane-3b, 17b-diol [3b-Adiol] inhibits prostate cancer cell migration through activation of the estrogen receptor beta subtype.�8 Other researchers reported that �3b-Adiol not only inhibits PC3-Luc cell [a specific type of prostate cancer cell] migratory properties, but also induces a broader antitumor phenotype [type of cell] by decreasing the proliferation [growth] rate, increasing cell adhesion [cancer cells don�t �stick� as normal cells do] and reducing invasive capabilities in vitro.�9 But these researchers went beyond test-tubes to living mice, writing �In vivo, continuous administration of 3b-Adiol reduces growth of established tumors and counteracts metastasis formation when PC3-Luc cells are engrafted subcutaneously in nude mice or are injected into the prostate.�
The conclusion to this research article was very encouraging: �Since 3b-Adiol has no androgenic activity, and cannot be converted to androgenic compounds, the effects here described entail a novel potential application of this agent against human PC.�9 A novel potential application of 3b-Adiol, a totally natural human testosterone, against human prostate cancer! Where are the headlines? This article was published in 2010!
For the really technically inclined, here are several �mechanisms of action� of 3b-Adiol, all of which come from stimulation of estrogen receptor beta:
- repression of VEGF-A (vascular endothelial growth factor A) expression
- destabilization of HIF-1a (hypoxia-inducible factor 1a)
- reduction of �Snail1″ relocation from the cytoplasm to the nucleus of cancer cells
According to the researchers who published the above mechanisms of action, �� high Gleason grade cancers � exhibit significantly more HIF-1a and VEGF-A and Snail1 nuclear localization compared to low Gleason grade cancers.�
The murine hair follicle undergoes pronounced cyclic expansion and regression, leading to rapidly changing demands for its vascular support. Our study aimed to quantify the cyclic changes of perifollicular vascularization and to characterize the biological role of VEGF for hair growth, angiogenesis, and follicle cycling. We found a significant increase in perifollicular vascularization during the growth phase (anagen) of the hair cycle, followed by regression of angiogenic blood vessels during the involution (catagen) and the resting (telogen) phase. Perifollicular angiogenesis was temporally and spatially correlated with upregulation of VEGF mRNA expression by follicular keratinocytes of the outer root sheath, but not by dermal papilla cells. Transgenic overexpression of VEGF in outer root sheath keratinocytes of hair follicles strongly induced perifollicular vascularization, resulting in accelerated hair regrowth after depilation and in increased size of hair follicles and hair shafts. Conversely, systemic treatment with a neutralizing anti-VEGF antibody led to hair growth retardation and reduced hair follicle size. No effects of VEGF treatment or VEGF blockade were observed in mouse vibrissa organ cultures, which lack a functional vascular system. These results identify VEGF as a major mediator of hair follicle growth and cycling and provide the first direct evidence that improved follicle vascularization promotes hair growth and increases hair follicle and hair size.

Heres a study demonstrating the role of hif-1 in VEGF regulation:
Transcriptional Regulation Controls Angiogenesis in Hypoxia
One important HIF-1 function is to promote angiogenesis; HIF-1 directs migration of mature endothelial cells toward a hypoxic environment [2,5]. This is done via HIF-1 regulation of vascular endothelial growth factor (VEGF) transcription. VEGF is a major regulator of angiogenesis, which promotes endothelial cell migration toward a hypoxic area. During hypoxia, HIF-1 binds the regulatory region of the VEGF gene, inducing its transcription and initiating its expression [12,15,16]. Such endothelial cells ultimately help to form new blood vessels, supplying the given area with oxygenated blood [14].
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2140184/
One important HIF-1 function is to promote angiogenesis; HIF-1 directs migration of mature endothelial cells toward a hypoxic environment [2,5]. This is done via HIF-1 regulation of vascular endothelial growth factor (VEGF) transcription. VEGF is a major regulator of angiogenesis, which promotes endothelial cell migration toward a hypoxic area. During hypoxia, HIF-1 binds the regulatory region of the VEGF gene, inducing its transcription and initiating its expression [12,15,16]. Such endothelial cells ultimately help to form new blood vessels, supplying the given area with oxygenated blood [14].
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2140184/
Androgens stimulate hypoxia-inducible factor 1 activation via autocrine loop of tyrosine kinase receptor/phosphatidylinositol 3'-kinase/protein kinase B in prostate cancer cells.
Dihydrotestosterone (DHT) activates HIF-1alpha nuclear protein expression in LNCaP cells but not in androgen receptor-negative PC-3 cells. HIF-1alpha expression is correlated with the transactivation of a hypoxia-responsive element-driven reporter gene and with the production of VEGF protein. The effect of DHT on HIF-1 was blocked by nonsteroidal antiandrogens, flutamide and bicalutamide. DHT does not affect HIF-1alpha mRNA levels but regulates HIF-1alpha protein expression through a translation-dependent pathway. PC-3 cells when incubated with increasing amounts of conditioned medium from LNCaP cells treated with DHT experienced a dose-dependent increase in HIF-1alpha. This induction was not seen either when LNCaP cells were treated with flutamide or conditioned medium were pretreated with antibody to the epidermal growth factor (EGF). HIF-1 activation by DHT was blocked by LY294002, a potent inhibitor of the phosphatidylinositol 3'-kinase signaling pathway, whereas HIF-1 activation by EGF, as ligand, was not inhibited by flutamide. In contrast, HIF-2alpha protein was not affected by androgens or antiandrogens.
Dihydrotestosterone (DHT) activates HIF-1alpha nuclear protein expression in LNCaP cells but not in androgen receptor-negative PC-3 cells. HIF-1alpha expression is correlated with the transactivation of a hypoxia-responsive element-driven reporter gene and with the production of VEGF protein. The effect of DHT on HIF-1 was blocked by nonsteroidal antiandrogens, flutamide and bicalutamide. DHT does not affect HIF-1alpha mRNA levels but regulates HIF-1alpha protein expression through a translation-dependent pathway. PC-3 cells when incubated with increasing amounts of conditioned medium from LNCaP cells treated with DHT experienced a dose-dependent increase in HIF-1alpha. This induction was not seen either when LNCaP cells were treated with flutamide or conditioned medium were pretreated with antibody to the epidermal growth factor (EGF). HIF-1 activation by DHT was blocked by LY294002, a potent inhibitor of the phosphatidylinositol 3'-kinase signaling pathway, whereas HIF-1 activation by EGF, as ligand, was not inhibited by flutamide. In contrast, HIF-2alpha protein was not affected by androgens or antiandrogens.
Evolution could be responsible for this DHT driven inhibition of androgen's procarcinogenic effects i.e the males with higher androgens were physically and mentally (androgen increases grey matter/spatial ability) stronger but were also susceptible to early cancer related deaths, so the males that ended up surviving were the ones that had the AR related edge and developed a feedback loop that canceled out the negative effects of androgens. Some of these biological feedback loops have been refined over thousands of years an as a result have become increasingly complex due to natural selection (survival of the fittest/most adaptable).
This is all interesting with regards to the prostate but what about the scalp?
First lets take a look at how exactly 3 beta diol is synthesized.
5α-Androstane-3β,17β-diol, also called 3β-androstanediol, and often shortened to 3β-diol, is an endogenous steroid hormone. It is a 5α-reduced and 17β-hydroxylated metabolite of dehydroepiandrosterone (DHEA) as well as a 3β-hydroxylated metabolite of dihydrotestosterone (DHT). Similarly to DHEA, 3β-diol is a high-affinity full agonist of the ERβ, and hence, an estrogen.
https://en.wikipedia.org/wiki/5%CE%B...,17%CE%B2-diol
https://en.wikipedia.org/wiki/5%CE%B...,17%CE%B2-diol
3 beta diol can be synthesized from either DHEA or DHT via the respective enzymes. An important bit of information to note is that both 3 beta diol and DHEA activate ERbeta.
Regional scalp differences of the androgenic metabolic pattern in subjects affected by male pattern baldness.
Regional differences in the androgen metabolism were established in alopecic and non alopecic areas of patients affected by male pattern baldness (MPB). 5-alpha-reductase (5-alpha-R) activity was measured by the formation of dihydrotestosterone (DHT), using 3H-testosterone as substrate: this activity was higher in the alopecic areas (3.4 pmol/g tissue/h) than in the non alopecic skin (1.5 pmol/g tissue/h). 3-alpha,beta-hydroxysteroid oxoreductase (3-alpha, beta-HO) was studied using 3H-DHT as precursor and measuring the corresponding formed 3-alpha- and 3-beta-androstanediols (alpha DIOL and beta DIOL). The beta DIOL was the predominant metabolite and total 3-alpha, beta-HO activity was higher in alopecic skin (12.4 pmol/g tissue/h) than in non alopecic areas (8.4 pmol/g tissue/h). Also 17, beta-hydroxysteroid oxoreductase was measured using either testosterone or DHT as substrates: androstenedione formed from testosterone was higher in hairy skin (12 pmol/g tissue/h) than in alopecic areas (6 pmol/g tissue/h); androstanedione formed from DHT was also higher in non alopecic areas (8.1 pmol/g tissue/h) than in alopecic skin (2.8 pmol/g tissue/h). The greater formation of beta DIOL in the sebaceous glands-enriched alopecic skin supports the hypothesis for a specific role of this metabolite in the control of the sebaceous activity.
Regional scalp differences of the androgenic metabolic pattern in subjects affected by male pattern baldness.
Regional differences in the androgen metabolism were established in alopecic and non alopecic areas of patients affected by male pattern baldness (MPB). 5-alpha-reductase (5-alpha-R) activity was measured by the formation of dihydrotestosterone (DHT), using 3H-testosterone as substrate: this activity was higher in the alopecic areas (3.4 pmol/g tissue/h) than in the non alopecic skin (1.5 pmol/g tissue/h). 3-alpha,beta-hydroxysteroid oxoreductase (3-alpha, beta-HO) was studied using 3H-DHT as precursor and measuring the corresponding formed 3-alpha- and 3-beta-androstanediols (alpha DIOL and beta DIOL). The beta DIOL was the predominant metabolite and total 3-alpha, beta-HO activity was higher in alopecic skin (12.4 pmol/g tissue/h) than in non alopecic areas (8.4 pmol/g tissue/h). Also 17, beta-hydroxysteroid oxoreductase was measured using either testosterone or DHT as substrates: androstenedione formed from testosterone was higher in hairy skin (12 pmol/g tissue/h) than in alopecic areas (6 pmol/g tissue/h); androstanedione formed from DHT was also higher in non alopecic areas (8.1 pmol/g tissue/h) than in alopecic skin (2.8 pmol/g tissue/h). The greater formation of beta DIOL in the sebaceous glands-enriched alopecic skin supports the hypothesis for a specific role of this metabolite in the control of the sebaceous activity.
When I read this I immediately recalled a compound that is commonly used for treating Androgenetic Alopecia: Ketoconazole (nizoral).
Ketoconazole is actually a really effective inhibitor of androgen synthesis, and a weak inhibitor of Androgen receptor.
In vitro inhibition by ketoconazole of human testicular steroid oxidoreductases.
An oral antimycotic agent, ketoconazole has been demonstrated to be an inhibitor of cytochrome P-450-dependent monooxygenases. To investigate its effect on steroid oxidoreductases, in vitro studies were carried out using subcellular fractions of human testes. Ketoconazole competitively inhibited activities of 3 beta-hydroxy-5-ene-steroid oxidoreductase/isomerase and NADH-linked 20 alpha-hydroxysteroid oxidoreductase for steroid substrate and the Ki values were 2.9 and 0.9 microM, respectively. In contrast, ketoconazole inhibited neither 17 beta-hydroxysteroid oxidoreductase nor NADPH-linked 20 alpha-hydroxysteroid oxidoreductase, indicating that the two 20 alpha-hydroxysteroid oxidoreductases are distinct. Further, ketoconazole inhibited non-competitively the above enzyme activities for the corresponding cofactors of NAD and NADH. From the binding mode of ketoconazole to cytochrome P-450 and the present findings, it appears likely that the agent binds to a site which is different from that of steroids or pyridine nucleotides.
An oral antimycotic agent, ketoconazole has been demonstrated to be an inhibitor of cytochrome P-450-dependent monooxygenases. To investigate its effect on steroid oxidoreductases, in vitro studies were carried out using subcellular fractions of human testes. Ketoconazole competitively inhibited activities of 3 beta-hydroxy-5-ene-steroid oxidoreductase/isomerase and NADH-linked 20 alpha-hydroxysteroid oxidoreductase for steroid substrate and the Ki values were 2.9 and 0.9 microM, respectively. In contrast, ketoconazole inhibited neither 17 beta-hydroxysteroid oxidoreductase nor NADPH-linked 20 alpha-hydroxysteroid oxidoreductase, indicating that the two 20 alpha-hydroxysteroid oxidoreductases are distinct. Further, ketoconazole inhibited non-competitively the above enzyme activities for the corresponding cofactors of NAD and NADH. From the binding mode of ketoconazole to cytochrome P-450 and the present findings, it appears likely that the agent binds to a site which is different from that of steroids or pyridine nucleotides.
Hydroxylation of 5 alpha-androstane-3 beta,17 beta-diol by rat prostate microsomes: potent inhibition by imidazole-type antimycotic drugs and lack of inhibition by steroid 5 alpha-reductase inhibitors.
5 alpha-Dihydrotestosterone, the principal androgen mediating prostate growth and function in the rat, is formed from testosterone by steroid 5 alpha-reductase. The inactivation of 5 alpha-dihydrotestosterone involves reversible reduction to 5 alpha-androstane-3 beta,17 beta-diol by 3 beta-hydroxysteroid oxidoreductase followed by 6 alpha-, 7 alpha-, or 7 beta-hydroxylation. 5 alpha-Androstane-3 beta,17 beta-diol hydroxylation represents the ultimate inactivation step of dihydrotestosterone in rat prostate and is apparently catalyzed by a single, high-affinity (Km approximately 0.5 microM) microsomal cytochrome P450 enzyme. The present studies were designed to determine if 5 alpha-androstane-3 beta,17 beta-diol hydroxylation by rat prostate microsomes is inhibited by agents that are known inhibitors of androgen-metabolizing enzymes. Inhibitors of steroid 5 alpha-reductase (4-azasteroid analogs; 10 microM) or inhibitors of 3 beta-hydroxysteroid oxidoreductase (trilostane, azastene, and cyanoketone; 10 microM) had no appreciable effect on the 6 alpha-, 7 alpha-, or 7 beta-hydroxylation of 5 alpha-androstane-3 beta,17 beta-diol (10 microM) by rat prostate microsomes. Imidazole-type antimycotic drugs (ketoconazole, clotrimazole, and miconazole; 0.1-10 microM) all markedly inhibited 5 alpha-androstane-3 beta,17 beta-diol hydroxylation in a concentration-dependent manner, whereas triazole-type antimycotic drugs (fluconazole and itraconazole; 0.1-10 microM) had no inhibitory effect. The rank order of inhibitory potency of the imidazole-type antimycotic drugs was miconazole greater than clotrimazole greater than ketoconazole. In the case of clotrimazole, the inhibition was shown to be competitive in nature, with a Ki of 0.03 microM. The imidazole-type antimycotic drugs [B]inhibited all three pathways of 5 alpha-androstane-3 beta,17 beta-diol hydroxylation to the same extent[B], which provides further evidence that, in rat prostate microsomes, a single cytochrome P450 enzyme catalyzes the 6 alpha-, 7 alpha-, and 7 beta-hydroxylation of 5 alpha-androstane-3 beta,17 beta-diol. These studies demonstrate that certain imidazole-type compounds are potent, competitive inhibitors of 5 alpha-androstane-3 beta,17 beta-diol hydroxylation by rat prostate microsomes, which is consistent with the effect of these antimycotic drugs on cytochrome P450 enzymes involved in the metabolism of other androgens and steroids.
5 alpha-Dihydrotestosterone, the principal androgen mediating prostate growth and function in the rat, is formed from testosterone by steroid 5 alpha-reductase. The inactivation of 5 alpha-dihydrotestosterone involves reversible reduction to 5 alpha-androstane-3 beta,17 beta-diol by 3 beta-hydroxysteroid oxidoreductase followed by 6 alpha-, 7 alpha-, or 7 beta-hydroxylation. 5 alpha-Androstane-3 beta,17 beta-diol hydroxylation represents the ultimate inactivation step of dihydrotestosterone in rat prostate and is apparently catalyzed by a single, high-affinity (Km approximately 0.5 microM) microsomal cytochrome P450 enzyme. The present studies were designed to determine if 5 alpha-androstane-3 beta,17 beta-diol hydroxylation by rat prostate microsomes is inhibited by agents that are known inhibitors of androgen-metabolizing enzymes. Inhibitors of steroid 5 alpha-reductase (4-azasteroid analogs; 10 microM) or inhibitors of 3 beta-hydroxysteroid oxidoreductase (trilostane, azastene, and cyanoketone; 10 microM) had no appreciable effect on the 6 alpha-, 7 alpha-, or 7 beta-hydroxylation of 5 alpha-androstane-3 beta,17 beta-diol (10 microM) by rat prostate microsomes. Imidazole-type antimycotic drugs (ketoconazole, clotrimazole, and miconazole; 0.1-10 microM) all markedly inhibited 5 alpha-androstane-3 beta,17 beta-diol hydroxylation in a concentration-dependent manner, whereas triazole-type antimycotic drugs (fluconazole and itraconazole; 0.1-10 microM) had no inhibitory effect. The rank order of inhibitory potency of the imidazole-type antimycotic drugs was miconazole greater than clotrimazole greater than ketoconazole. In the case of clotrimazole, the inhibition was shown to be competitive in nature, with a Ki of 0.03 microM. The imidazole-type antimycotic drugs [B]inhibited all three pathways of 5 alpha-androstane-3 beta,17 beta-diol hydroxylation to the same extent[B], which provides further evidence that, in rat prostate microsomes, a single cytochrome P450 enzyme catalyzes the 6 alpha-, 7 alpha-, and 7 beta-hydroxylation of 5 alpha-androstane-3 beta,17 beta-diol. These studies demonstrate that certain imidazole-type compounds are potent, competitive inhibitors of 5 alpha-androstane-3 beta,17 beta-diol hydroxylation by rat prostate microsomes, which is consistent with the effect of these antimycotic drugs on cytochrome P450 enzymes involved in the metabolism of other androgens and steroids.
Ketoconazole/miconzole inhibit 3 Beta HSD
3 beta HSD converts DHT to 3beta diol.
3beta diol activates ERbeta.
ERbeta destabilizes HIF-1 and results in loss of VEGF expression.
Hair follicles fail to enter anagen due to lack of bood supply.
This is a potential explanation of the mechanism behind ketoconazoles hair growth/hair loss prevention effects.
I also wanted to talk a bit about PGD2. Specifically it�s metabolite:
15-dPGJ2 and PGD2 inhibit hair growth in mouse and human hair follicles
Given the temporal peak of PGD2 before the apoptotic catagen stage, the published ability of its metabolite 15-dPGJ2 to induce apoptosis in other cell types, we tested the effects of the prostaglandins on primary cell culture of keratinocytes isolated from neonatal foreskin. 15-dPGJ2 induces apoptosis (fig. S2A), as evidenced by plasma membrane blebbing and cell retraction/shrinkage. 15-dPGJ2 also decreased cell density, cell division, and live-cell numbers (fig. S2, B to D). Perhaps because the origin of these keratinocytes was not the hair follicle, PGD2 had no such effect on the cells. However, 15-dPGJ2 did increase sub-G1 DNA quantities and activated caspase 3 in human keratinocytes, which are features of apoptotic cell death (fig. S2, E to G). We therefore hypothesized that at least 15-dPGJ2, if not also PGD2, could directly inhibit hair growth in vivo.
15-dPGJ2 was applied topically to dorsal back skin of C57BL/6 mice that had been depilated to synchronize the hair follicle cycle. Starting on day 8 after depilation and continuing every other day, we applied 10 μg of 15-dPGJ2 or acetone vehicle. Hair length was measured on days 4, 12, 14, and 16 after depilation. On days 12 to 16, hair at the site of treatment was shorter than in vehicle-treated animals (Fig. 6A). To determine a minimal effective dose, we tested the application of 1 μg of both PGD2 and 15-dPGJ2 as above and measured hair length on day 20 after depilation. PGD2 inhibited hair growth, but to a lesser extent than 15-dPGJ2 (Fig. 6B). We found no evidence of changes in hair follicle cycling grossly or by histologic examination.
Given the temporal peak of PGD2 before the apoptotic catagen stage, the published ability of its metabolite 15-dPGJ2 to induce apoptosis in other cell types, we tested the effects of the prostaglandins on primary cell culture of keratinocytes isolated from neonatal foreskin. 15-dPGJ2 induces apoptosis (fig. S2A), as evidenced by plasma membrane blebbing and cell retraction/shrinkage. 15-dPGJ2 also decreased cell density, cell division, and live-cell numbers (fig. S2, B to D). Perhaps because the origin of these keratinocytes was not the hair follicle, PGD2 had no such effect on the cells. However, 15-dPGJ2 did increase sub-G1 DNA quantities and activated caspase 3 in human keratinocytes, which are features of apoptotic cell death (fig. S2, E to G). We therefore hypothesized that at least 15-dPGJ2, if not also PGD2, could directly inhibit hair growth in vivo.
15-dPGJ2 was applied topically to dorsal back skin of C57BL/6 mice that had been depilated to synchronize the hair follicle cycle. Starting on day 8 after depilation and continuing every other day, we applied 10 μg of 15-dPGJ2 or acetone vehicle. Hair length was measured on days 4, 12, 14, and 16 after depilation. On days 12 to 16, hair at the site of treatment was shorter than in vehicle-treated animals (Fig. 6A). To determine a minimal effective dose, we tested the application of 1 μg of both PGD2 and 15-dPGJ2 as above and measured hair length on day 20 after depilation. PGD2 inhibited hair growth, but to a lesser extent than 15-dPGJ2 (Fig. 6B). We found no evidence of changes in hair follicle cycling grossly or by histologic examination.
I�m still doing research on this by product of PGD2 as it may hold more relevance than PGD2 itself.
Another very crucial effect of DHT is the elevation Dickkopf-1 (DKK1).
Dihydrotestosterone-inducible dickkopf 1 from balding dermal papilla cells causes apoptosis in follicular keratinocytes.
Abstract
Recent studies suggest that androgen-driven alteration to the autocrine and paracrine factors produced by scalp dermal papilla (DP) cells may be a key to androgen-potentiated balding. Here, we screened dihydrotestosterone (DHT)-regulated genes in balding DP cells and found that dickkopf 1 (DKK-1) is one of the most upregulated genes. DKK-1 messenger RNA is upregulated in 3-6 hours after 50-100 nM DHT treatment and ELISA showed that DKK-1 is secreted from DP cells in response to DHT. A co-culture system using outer root sheath (ORS) keratinocytes and DP cells showed that DHT inhibits the growth of ORS cells, and neutralizing antibody against DKK-1 significantly reversed the growth inhibition of ORS cells. Analysis of co-cultured ORS cells showed a significant increment of sub-G1 apoptotic cells in response to DHT. Also, recombinant human DKK-1 inhibited the growth of ORS cells and triggered apoptotic cell death. In addition, DHT-induced epithelial cell death in cultured hair follicles was reversed by neutralizing DKK-1 antibody. Moreover, immunoblotting showed that the DKK-1 level is up in the bald scalp compared with the haired scalp of patients with androgenetic alopecia. Altogether, our data strongly suggest that DHT-inducible DKK-1 is involved in DHT-driven balding.
Abstract
Recent studies suggest that androgen-driven alteration to the autocrine and paracrine factors produced by scalp dermal papilla (DP) cells may be a key to androgen-potentiated balding. Here, we screened dihydrotestosterone (DHT)-regulated genes in balding DP cells and found that dickkopf 1 (DKK-1) is one of the most upregulated genes. DKK-1 messenger RNA is upregulated in 3-6 hours after 50-100 nM DHT treatment and ELISA showed that DKK-1 is secreted from DP cells in response to DHT. A co-culture system using outer root sheath (ORS) keratinocytes and DP cells showed that DHT inhibits the growth of ORS cells, and neutralizing antibody against DKK-1 significantly reversed the growth inhibition of ORS cells. Analysis of co-cultured ORS cells showed a significant increment of sub-G1 apoptotic cells in response to DHT. Also, recombinant human DKK-1 inhibited the growth of ORS cells and triggered apoptotic cell death. In addition, DHT-induced epithelial cell death in cultured hair follicles was reversed by neutralizing DKK-1 antibody. Moreover, immunoblotting showed that the DKK-1 level is up in the bald scalp compared with the haired scalp of patients with androgenetic alopecia. Altogether, our data strongly suggest that DHT-inducible DKK-1 is involved in DHT-driven balding.
DKK1 is a high affinity antagonistic ligand for LRP6, which is a Wnt coreceptor that acts together with the Frizzled serpentine receptor to initiate Wnt signal transduction. Two different models have been proposed to account for the mechanism by which DKK1 antagonizes LRP6 function. One model suggests that DKK1 binding to LRP6 disrupts Wnt-induced Frizzled-LRP6 complex formation, whereas the other model proposes that DKK1 interaction with LRP6 promotes LRP6 internalization and degradation, thereby reducing the cell surface LRP6 level.
WNT - Canonical pathway
The canonical Wnt pathway (or Wnt/β-catenin pathway) is the Wnt pathway that causes an accumulation of β-catenin in the cytoplasm and its eventual translocation into the nucleus to act as a transcriptional coactivator of transcription factors that belong to the TCF/LEF family. Without Wnt signaling, the β-catenin would not accumulate in the cytoplasm since a destruction complex would normally degrade it. This destruction complex includes the following proteins: Axin, adenomatosis polyposis coli (APC), protein phosphatase 2A (PP2A), glycogen synthase kinase 3 (GSK3) and casein kinase 1α (CK1α).[14] It degrades β-catenin by targeting it forubiquitination, which subsequently sends it to the proteasome to be digested.[11][15] However, as soon as Wnt binds Fz and LRP5/6, the destruction complex function becomes disrupted. This is due to Wnt causing the translocation of the negative Wnt regulator, Axin, and the destruction complex to the plasma membrane.Phosphorylation by other proteins in the destruction complex subsequently binds Axin to the cytoplasmic tail of LRP5/6. Axin becomes de-phosphorylated and its stability and levels are decreased. Dsh then becomes activated via phosphorylation and its DIX and PDZ domains inhibit the GSK3 activity of the destruction complex. This allows β-catenin to accumulate and localize to the nucleus and subsequently induce a cellular response via gene transduction alongside the TCF/LEF (T-cell factor/lymphoid enhancing factor)[16]transcription factors.[15]
The canonical Wnt pathway (or Wnt/β-catenin pathway) is the Wnt pathway that causes an accumulation of β-catenin in the cytoplasm and its eventual translocation into the nucleus to act as a transcriptional coactivator of transcription factors that belong to the TCF/LEF family. Without Wnt signaling, the β-catenin would not accumulate in the cytoplasm since a destruction complex would normally degrade it. This destruction complex includes the following proteins: Axin, adenomatosis polyposis coli (APC), protein phosphatase 2A (PP2A), glycogen synthase kinase 3 (GSK3) and casein kinase 1α (CK1α).[14] It degrades β-catenin by targeting it forubiquitination, which subsequently sends it to the proteasome to be digested.[11][15] However, as soon as Wnt binds Fz and LRP5/6, the destruction complex function becomes disrupted. This is due to Wnt causing the translocation of the negative Wnt regulator, Axin, and the destruction complex to the plasma membrane.Phosphorylation by other proteins in the destruction complex subsequently binds Axin to the cytoplasmic tail of LRP5/6. Axin becomes de-phosphorylated and its stability and levels are decreased. Dsh then becomes activated via phosphorylation and its DIX and PDZ domains inhibit the GSK3 activity of the destruction complex. This allows β-catenin to accumulate and localize to the nucleus and subsequently induce a cellular response via gene transduction alongside the TCF/LEF (T-cell factor/lymphoid enhancing factor)[16]transcription factors.[15]
It�s very clear that without Beta Catenin hair will not form:
Further analysis demonstrates that β-catenin is essential for fate decisions of skin stem cells: in the absence of β-catenin, stem cells fail to differentiate into follicular keratinocytes, but instead adopt an epidermal fate.
I�ve come across mainly dead ends and no studies to date actually document how Dkk1 is regulated.
We know that L-ascorbic acid 2-phosphate and L-threonate, an ascorbate metabolite inhibits DHT induced DKK-1.
Dkk-1 is also induced by p53: http://www.ncbi.nlm.nih.gov/pubmed/10777218
DHT increases p53 and p21 significantly: http://www.ncbi.nlm.nih.gov/pubmed/22859066
This is a weak connection at best.
Other possible links include:
The Wnt antagonist DICKKOPF-1 gene is induced by 1a,25-dihydroxyvitamin D3
The Wnt�b-catenin pathway is aberrantly activated in most colon cancers. DICKKOPF-1 (DKK-1) gene encodes an extracellular Wnt inhibitor that blocks the formation of signalling receptor complexes at the plasma membrane. We report that 1a,25- dihydroxyvitamin D3 [1,25(OH)2D3], the most active vitamin D metabolite, increases the level of DKK-1 RNA and protein in human SW480-ADH colon cancer cells. This effect is dose dependent, slow and depends on the presence of a transcription-competent nuclear vitamin D receptor (VDR). Accordingly, 1,25(OH)2D3 activates a 2300 bp fragment of the human DKK-1 gene promoter. Chromatin immunoprecipitation assays revealed that 1,25(OH)2D3 treatment induced a pattern of histone modifications which is compatible with transcriptionally active chromatin. Exogenous expression of E-cadherin into SW480-ADH cells results in a strong adhesive phenotype and a 17-fold increase in DKK-1 RNA. In contrast, an E-cadherin blocking antibody inhibits 1,25(OH)2D3-induced differentiation of SW480-ADH cells and DKK-1 gene expression. Remarkably, in vivo treatment with the vitamin D analogue EB1089 induced DKK-1 protein expression in SW480-ADH cells xenografted in immunodeficient mice, and a correlation was observed in the expression of VDR and DKK-1 RNA in a series of 32 human colorectal tumours. These data indicate that 1,25(OH)2D3 activates the transcription of the DKK-1 gene, probably in an indirect way that is associated to the promotion of a differentiated phenotype. DKK-1 gene induction constitutes a novel mechanism of inhibition of Wnt signalling and antitumour action by 1,25(OH)2D3.
The Wnt�b-catenin pathway is aberrantly activated in most colon cancers. DICKKOPF-1 (DKK-1) gene encodes an extracellular Wnt inhibitor that blocks the formation of signalling receptor complexes at the plasma membrane. We report that 1a,25- dihydroxyvitamin D3 [1,25(OH)2D3], the most active vitamin D metabolite, increases the level of DKK-1 RNA and protein in human SW480-ADH colon cancer cells. This effect is dose dependent, slow and depends on the presence of a transcription-competent nuclear vitamin D receptor (VDR). Accordingly, 1,25(OH)2D3 activates a 2300 bp fragment of the human DKK-1 gene promoter. Chromatin immunoprecipitation assays revealed that 1,25(OH)2D3 treatment induced a pattern of histone modifications which is compatible with transcriptionally active chromatin. Exogenous expression of E-cadherin into SW480-ADH cells results in a strong adhesive phenotype and a 17-fold increase in DKK-1 RNA. In contrast, an E-cadherin blocking antibody inhibits 1,25(OH)2D3-induced differentiation of SW480-ADH cells and DKK-1 gene expression. Remarkably, in vivo treatment with the vitamin D analogue EB1089 induced DKK-1 protein expression in SW480-ADH cells xenografted in immunodeficient mice, and a correlation was observed in the expression of VDR and DKK-1 RNA in a series of 32 human colorectal tumours. These data indicate that 1,25(OH)2D3 activates the transcription of the DKK-1 gene, probably in an indirect way that is associated to the promotion of a differentiated phenotype. DKK-1 gene induction constitutes a novel mechanism of inhibition of Wnt signalling and antitumour action by 1,25(OH)2D3.
Furthermore, DHT has been shown to upregulate VDR via - wait for it � ERBeta!
Sex steroids induced up-regulation of 1,25-(OH)2 vitamin D3 receptors in T 47D breast cancer cells.
Abstract
There is evidence indicating that 1,25-dihydroxyvitamin D3 [1,25(OH)2D3] through binding to its specific receptor (VDR) exerts an antiproliferative effect on breast cancer cells. Considering the importance of receptor regulation in modulating the target cell responsiveness to hormones, the effect of dihydrotestosterone (DHT) and estradiol-17 beta (E2) on the regulation of VDR number was investigated in T 47D human breast cancer cells that also express androgen and estrogen (ER) receptors. Exposure to 10(-7) M DHT for 72 h resulted in a significant increase in VDR levels. Similar results were obtained with 10(-7) M E2. DHT- and E2-induced up-regulation was completely suppressed by 10(-6) M tamoxifen (TAM) addition but unaffected by 10(-6) M flutamide. TAM treatment alone produced a significant dose-dependent increase in VDR content, that was maximal at 10(-6) M. Our data strongly suggest, for the first time, an up-regulation of VDR by DHT and E2 via an ER-mediated mechanism.
Abstract
There is evidence indicating that 1,25-dihydroxyvitamin D3 [1,25(OH)2D3] through binding to its specific receptor (VDR) exerts an antiproliferative effect on breast cancer cells. Considering the importance of receptor regulation in modulating the target cell responsiveness to hormones, the effect of dihydrotestosterone (DHT) and estradiol-17 beta (E2) on the regulation of VDR number was investigated in T 47D human breast cancer cells that also express androgen and estrogen (ER) receptors. Exposure to 10(-7) M DHT for 72 h resulted in a significant increase in VDR levels. Similar results were obtained with 10(-7) M E2. DHT- and E2-induced up-regulation was completely suppressed by 10(-6) M tamoxifen (TAM) addition but unaffected by 10(-6) M flutamide. TAM treatment alone produced a significant dose-dependent increase in VDR content, that was maximal at 10(-6) M. Our data strongly suggest, for the first time, an up-regulation of VDR by DHT and E2 via an ER-mediated mechanism.
Vitamin D has been shown to have a biphasic effect on hair growth:
At higher concentrations of 1,25(OH)2D3, there was a dose-dependent inhibition of both follicle and fiber growth (IC50 values of 100 nM), in part due to reduction in the growth periods. There was a marked delay between the onset of 1,25(OH)2D-induced hair follicle and hair fiber growth inhibition.
Time-course and dose-curve experiments showed that 1,25(OH)2D3 (107 M) caused a slow 3- to 5-fold induction of DKK-1 RNA at 24�48 h upon treatment. The effect of 1,25(OH)2D3 was specific, as several hormones (dexamethasone, retinoic acid, progesterone and oestradiol) acting through members of the superfamily of nuclear receptors similar to VDR did not induce DKK-1. The induction of DKK-1 was confirmed at the protein level and in another colon cancer cell line. Immunofluorescence studies confirmed the increase in DKK-1 protein expression following 1,25(OH)2D3 exposure and showed its preferential localization in the cell periphery, Golgi apparatus and vesicles of the exocytic route. These results confirmed that 1,25(OH)2D3 induces DKK-1 expression with slow kinetics, which precluded the use of translation inhibitors such as cycloheximide to investigate whether the induction is direct or indirect.
I strongly believe that inhibiting DKK-1 or reducing it will enable other treatments to work far more effectively, along with mitigating the detrimental effects of downstream DHT pathways i.e the actual effect instead of the overall mediator. The answer lies in the details, we must fully understand the mechanisms � the how and why before trying to exploit the pathway for our own gain. This is the key to hacking and efficient problem solving, you find the weakest link and plan your attack on that.
In the next post I will discuss therapeutic applications of these findings and some experiments I�ve been conducting (with interesting results), after all, what good is theory without application?













Comment